
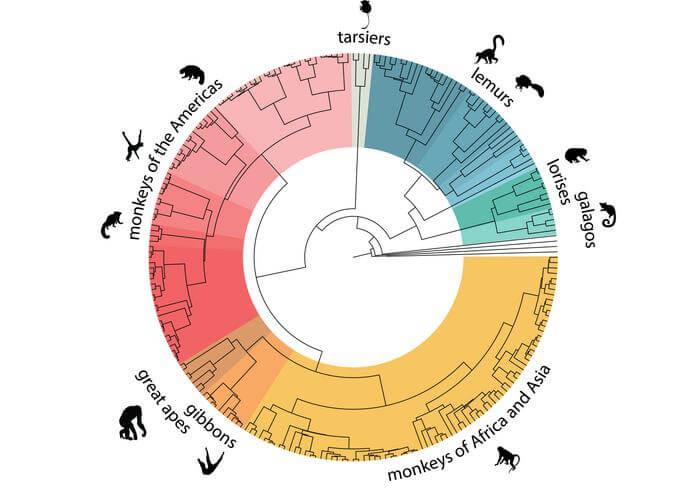
BARCELONA, Spain — Scientists may be closing in on the genetic origins of many modern-day diseases. A team has mapped the DNA of over 800 individuals from 233 primate species by studying the fossilized remains of our primitive ancestors. Nearly half of them are still in existence today. This substantial undertaking has quadrupled the number of completed genomes, thereby yielding fresh insights into the world’s biological diversity.
“Humans are primates. The study of hundreds of nonhuman primate genomes, given their phylogenetic position, is very valuable for human evolutionary studies, to better understand the human genome and the bases of our singularity, including the bases of human diseases, and for their future conservation,” says the lead author of the study, Professor Tomàs Marquès-Bonet from Pompeu Fabra University, in a media release.
At present, the causes of numerous prevalent diseases, such as diabetes and heart disease, remain uncertain. This is either due to insufficient genetic data or the complex interaction of many contributing factors. Some diseases are hypothesized to originate from a combination of genetic variations or mutations with subtle effects that, when acting together, lead to conditions like diabetes or cancer.
“6% of the 4.3 million missense mutations identified are abundant in primates and are therefore considered ‘potentially benign’ in human disease, given that their presence is tolerated in these animals” says the study’s co-author, Dr. Kyle Farh, Vice President of Artificial Intelligence at Illumina.
Missense mutations refer to DNA changes that lead to the encoding of different amino acids at a specific position in the resulting proteins. The multinational team utilized a deep learning algorithm named PrimateAI-3D, developed by Illumina, San Diego, a global leader in DNA sequencing. This innovative tool is akin to a chatbot, using genome sequences instead of human language.
(credit: Credit: Lukas Kuderna)
The resulting project provides the most comprehensive catalog of primate genomic data to date, featuring information on species from Asia, America, Africa, and Madagascar.
These studies indicate that primate genetics do not always align with their taxonomy, adds Prof. Marques-Bonet. The team discovered several instances where the relationships among primate species are best described as complex, network-like, rather than simple branching trees.
The research offers new insights into the evolution of baboons — a diverse and abundant group of monkeys. It reveals previously unknown instances of hybridization and gene flow among species. For example, the western Tanzanian yellow baboons are the first identified non-human primates to have received genetic input from three different lineages.
“These results suggest that the population genetic structure and history of introgression among baboon lineages is more complex than was previously thought, and that shows that the baboons are a good model for the evolution of humans, Neanderthals and Denisovans,” says co-lead author Prof. Jeffrey Rogers, from Baylor College of Medicine in Houston, Texas.
Neanderthals and Denisovans — a group named after the Siberian caves where their remains were found — are believed to have interbred with early modern humans. All three species coexisted on Earth approximately 50,000 years ago.
“Our studies provide clues as to which species are in most dire need of conservation efforts, and could help to identify the most effective strategies to preserve them,” adds Lukas Kuderna, the study’s first author and a Ph.D. student at Illumina.
This extensive catalog has also halved the number of genomic innovations previously believed to be exclusive to humans, facilitating the identification of mutations not shared with other primates that could be unique to the human species.
The study is published in the journal Science.
Scientists completed the human genome last year
In 2022, scientists successfully mapped the complete human genome. Researchers say it contains over six billion “letters” of DNA.
In 2003, scientists published a revised draft which was still missing more than eight percent on the genome. The hard-to-sequence long stretches were the most complex, full of repeating letters. They were impossible to decipher with the technology at the time. Two decades later, an international team finally has.
“In the future, when someone has their genome sequenced, we will be able to identify all of the variants in their DNA and use that information to better guide their healthcare,” says lead author Dr. Adam Phillippy from the University of Maryland in a media release.
South West News Service writer Mark Waghorn contributed to this report.
