(Credit: Karolina Grabowska from Pexels)
LOS ANGELES — Deafness has the reputation of often being an irreversible condition, as the unique sensory hearing cells of the inner ear that facilitate human hearing do not regenerate. Exciting new research out of Los Angeles, however, suggests modern medicine may one day be able to develop regenerative hearing treatments that could cure deafness.
Across two new studies, both partially funded by the National Institutes of Health, University of Southern California stem cell scientists explain why our sense of hearing is so delicate and how researchers can create new hearing treatments using these cells.
“In the non-sensory supporting cells of the inner ear, key genes required for conversion to sensory cells are shut off through a process known as ‘epigenetic silencing.’ By studying how the genes are shut off, we begin to understand how we might turn them back on to regenerate hearing,” says John Duc Nguyen, the first author of one of the papers, in a university release.
Nguyen now works at the biotech company Genentech and recently earned his PhD in the USC Stem Cell laboratory of Neil Segil, who passed away from pancreatic cancer in 2022. The second study explored when and how the ability to form sensory hearing cells is developed in the inner ear in the first place, noting and describing two specific genes that may be useful for regenerating hearing in adults.
“We focused on the genes Sox4 and Sox11 because we found that they are necessary for forming sensory hearing cells during development,” explains the paper’s first author Emily Xizi Wang, who also conducted her research as a PhD student in the Segil Lab and works at the biotech company Atara Biotherapeutics.
“These two papers are not only great science, but also a clear example of Neil Segil’s enduring legacy as an exceptional mentor to the next generation of stem cell researchers,” adds Gage Crump, a co-author on both papers and the interim chair of USC’s Department of Stem Cell Biology and Regenerative Medicine at the Keck School of Medicine of USC.
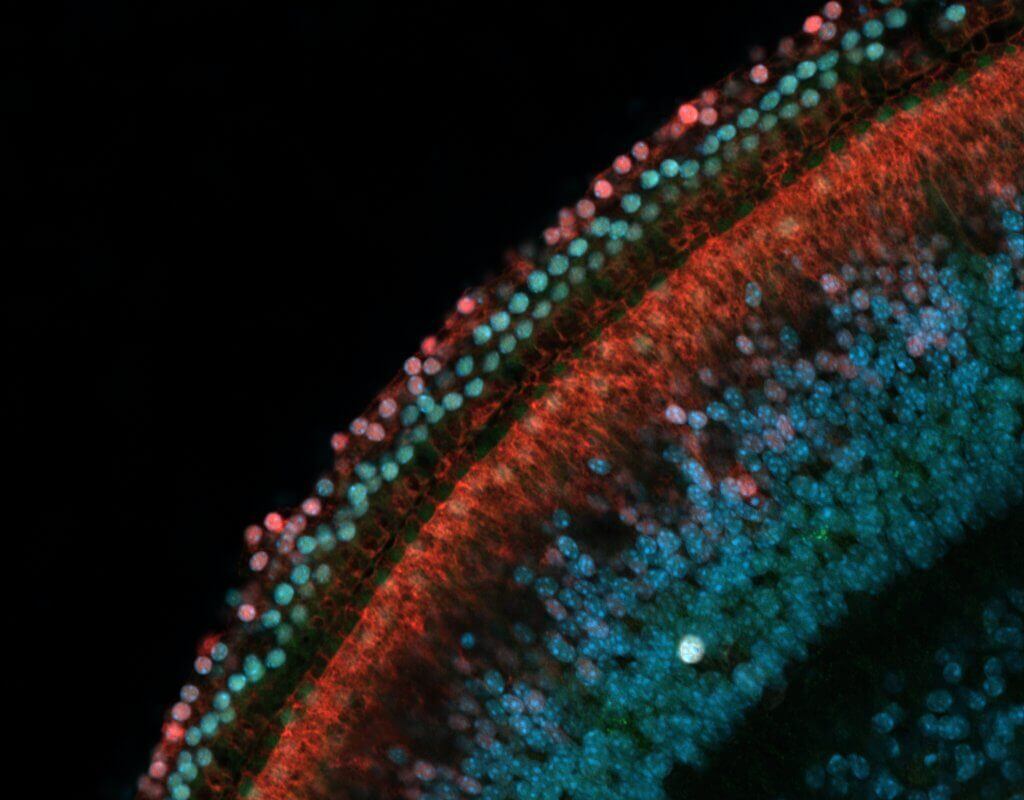
One key way that genes shut off or are “silenced” involves chemical compounds called methyl groups that bind to DNA, making it inaccessible. This topic is the focus of Nguyen’s paper. In other words, when DNA in charge of instructing a cell to become a sensory hearing cell is methylated, the cell can’t access those instructions.
Through experiments conducted with non-sensory supporting cells extracted from the inner ears of mice, Nguyen and a team were able to confirm that DNA methylation indeed silences genes known to promote conversion into sensory hearing cells. More specifically, the gene Atoh1 is considered a “master regulator” of inner ear development.
Importantly, an enzyme called TET can remove methyl groups from the DNA, consequently reversing the gene silencing and restoring the capability of supporting cells to turn into sensory hair cells. So, when researchers blocked the activity of TET, the supporting cells retained their DNA methylation – and thus could not convert into sensory hair cells in the Petri dish.
During a separate experiment, the research team tested the extent of gene silencing in supporting cells from a chronically deafened mouse. This approach led to the discovery that gene silencing was partially reversed, indicating that supporting cells once again had the capacity to respond to signals to transform into sensory hearing cells.
These results hold major implications; the loss of sensory hearing cells itself might partially reverse gene silencing in supporting cells in chronically deaf individuals. If that’s true, study authors explain the supporting cells of chronically deaf individuals may already be “naturally primed” to convert into sensory hearing cells.

For the second study, Wang and her colleagues analyzed when and how progenitor cells found in the inner ear form the capacity to form sensory hearing cells. They pinpointed when progenitor cells acquire this ability; between days 12 and 13.5 of embryonic development in mice. During that window of time, progenitor cells acquire the ability to respond to signals from the master regulator gene Atoh1 that trigger the formation of sensory hearing cells later on in development.
What primes the progenitor cells to respond to Atoh1? A pair of additional genes, Sox4 and Sox11, that change the state of these cells.
Among embryonic mice lacking Sox4 and Sox11, progenitor cells in the inner ear fail to develop into sensory hearing cells. Specifically, a loss of Sox4 and Sox11 made their cells’ DNA inaccessible. That effect is quite similar to DNA methylation. With the DNA inaccessible, the progenitor cells can’t respond to signals from Atoh1.
However, high levels of Sox4 and Sox11 activity stimulated mouse progenitor cells and supporting cells to form sensory hearing cells in a Petri dish setting.
On a promising note, in mice with damaged sensory cells in the inner ear, high levels of Sox4 and Sox11 activity seemed to increase the percentage of vestibular supporting cells converting into sensory receptor cells (from 6% to 40%).
“We’re excited to continue exploring the mechanisms by which cells in the inner ear gain the ability to differentiate as sensory cells during development and how these can be used to promote the recovery of sensory hearing cells in the mature inner ear,” concludes the paper’s corresponding author Ksenia Gnedeva.
Gnedeva is now an assistant professor in the USC Tina and Rick Caruso Department of Otolaryngology – Head and Neck Surgery, and the Department of Stem Cell Biology and Regenerative Medicine.
The studies are published in the Proceedings of the National Academy of Sciences.








I do suffer with Profound hearing loss in both ears and need a solution. Can a time frame be earmark for drugs on the market to assist in reversing hearing loss
I would like to be in on the study . I am currently on my 3rd set of aids. I am 76 years old, diabetic and in very good health.
Count me in on the research
Another possible volunteer for this treatment. Clinical trials etc. Is there a way to make it official connection to the researchers?
It’s not until you have hearing loss that you realize what a disadvantage it is. Mine is partly due to age and partly because of a condition known as menaries brought I’m told by stress which also affects balance. Due to my wife’s death from cancer I now live alone but have joined interest groups so as to not become too isolated. I have become a master at listening to conversations and looking as if I comprehend when actually I don’t. Hearing on phones is a nightmare as many voices don’t translate clearly. WhatsApp can work quite well. I agree, a Nobel prize would be well deserved.
I really will be happy if this stem cells can be realesed. as I’ve stopped using my hearing aids since they make me worse and could not hear properly to people everytime. So as me in this if stem cells are found
Sounds a lot better than hearing aids!
What stem cells are required, and How are they obtained.? My hearing has declined significantly in last 12 months , so could benefit directly, if this is a valid method of repair.! L.Kavanagh.B.Sc.
The scientists who be able to give natural hearing to deafs, will be awarded with Noble prize.
I personally been suffering from Ski slop high frequency hearing loss. The best hearing aids can only help to limit. Hopefully, Stem cell operations will be better and permanent solution. I don’t mind to leave my name and contact details to this Department, for to use my ears for further research and experiments. Hope their Department shall contact me.
I have healthy looking eardrums but I only have a small percentage of hearing. I would love to be a candidate of stem cell therapy or research.
This is great news.i hve been waiting for along time for a breakthrough in research to deafness.with modern technology anything is possible.i suffer from tinnitus n hearing problems so I can’t wait for someone to discover something that will help deaf people.there is nothing worse than not hearing a proper conversation or missing words .there is also a stigma attached to it as well.